Analyse the active site of an enzymatic domain#
This example is adapted from the method used by AntiSMASH to annotate biosynthetic gene clusters. AntiSMASH uses profile HMMs to annotate enzymatic domains in protein sequences. By matching the amino acids in the alignment, it can then predict the product specificity of the enzyme.
In this notebook, we show how to reproduce this kind of analysis, using a PKSI Acyltransferase domain built by the AntiSMASH authors (the HMM in HMMER2 format can be downloaded from their git repository).
References
[1]:
import pyhmmer
pyhmmer.__version__
[1]:
'0.12.1'
Loading the HMM#
Let’s start by downloading the PSKI-AT HMM from the AntiSMASH repository. We can use urllib.request to do that from within our Python script:
[2]:
import shutil
from urllib.request import urlopen
with urlopen('https://raw.githubusercontent.com/antismash/antismash/refs/heads/8-0-stable/antismash/modules/active_site_finder/data/PKSI-AT.hmm2') as src:
with open("PKSI-AT.hmm2", "wb") as dst:
shutil.copyfileobj(src, dst)
Loading a HMMER profile is done with the pyhmmer.plan7.HMMFile class, which provides an iterator over the HMMs in the file. We can use the read method to get the first (and only) pyhmmer.plan7.HMM from the file.
[3]:
with pyhmmer.plan7.HMMFile("PKSI-AT.hmm2") as hmm_file:
hmm = hmm_file.read()
Loading digitized sequences#
Easel provides the code necessary to load sequences from files in common biological formats, such as GenBank or FASTA. These utilities are wrapped by the pyhmmer.easel.SequenceFile, which provides an iterator over the sequences in the file. Note that SequenceFile tries to guess the format by default, but you can force a particular format with the format keyword argument. Note that we use digital=True to instruct the sequence file that we want to load sequences from the file into
digital format. The alphabet will be guessed from the sequence content, unless given explicitly with the alphabet keyword.
[4]:
with pyhmmer.easel.SequenceFile("data/seqs/PKSI.faa", digital=True) as seq_file:
sequences = seq_file.read_block()
Note
The C interface of Easel allows storing a sequence in two different modes: in text mode, where the sequence letters are represented as individual characters (e.g. “A” or “Y”), and digital mode, where sequence letters are encoded as digits. To make Python programs clearer, and to allow static typecheck of the storage mode, we provide two separate classes, TextSequence and DigitalSequence, that represent a sequence stored in either of these modes. Most functions that perform actual
work (such as pyhmmer.hmmsearch) will expect a digital sequence.
Running a search pipeline#
With the sequences and the HMM ready, we can finally run the search pipeline: it has to be initialized with an Alphabet instance, so that the Plan7 background model can be configured accordingly. Then, we run the pipeline in search mode, providing it one HMM, and several sequences. This method returns a TopHits instance that is already sorted and thresholded.
Note
Using a Pipeline object directly is fine when you only have a single HMM to compare to a sequence database, and when your sequence database is already stored in a supported type (a SequenceFile or a DigitalSequenceBlock). However, if you plan to make many-to-many comparisons between several sequences and several pHMMs, you should use the pyhmmer.hmmer.hmmsearch function, which will take care of setting up the multithreading and let you compute results efficiently on a multi-core
machine.
[5]:
pipeline = pyhmmer.plan7.Pipeline(hmm.alphabet)
hits = pipeline.search_hmm(hmm, sequences)
Rendering the alignments#
Domain instances store all the required information to report results in their alignment attribute. We can show the alignment between a HMM and a sequence like hmmsearch would as follow (using the first domain of the first hit as an example):
[6]:
ali = hits[0].domains[0].alignment
print(ali)
PKS-AT.tcoffee 1 lFpGQGsQyaGMGreLYetePVFRqalDrCaaaLrphLgfsLlevLfgdegqeeaaaslLdqTryaQPALFAvEYALArLWrSWGvePdAVlGHSvGEyvAAcvAGVlSLEDALrLVaaRGrLMqa.lpggGaMlaVraseeevrelLapyggrlsiAAvNGPrsvVvSGdaeaieallaeLeaqGirarrLkVsHAFHSplMepmldeleevlagitpraPriPliSnvTGewltgeealdpaYWarhlRePVrFadgletLlaelGctvFlEvGPhpvLtalarrtlgesagtngadaawlaSLrrg 308
+FpGQG+Q+aGMG eL++++ VF++a+ +C+aaL+p++++sL +v ++ +g a+ L++++++QP+ FAv+++LAr W+ Gv+P+AV+GHS+GE++AA+vAG+lSL+DA+r+V R++ ++a l+g+G+Ml+ ++se+ v e+La+++ +ls+AAvNGP ++VvSGd+ +ie+l++++ea G+rar ++V++A+HS+++e + el+evlag++p+aPr+P++S++ G+w+t+ +ld++YW+r+lR+ V Fa+++etL+ + G+t+F+Ev++hpvLt ++ t + la+Lrr+
sp|Q9ZGI5|PIKA1_STRVZ 635 VFPGQGTQWAGMGAELLDSSAVFAAAMAECEAALSPYVDWSLEAVVRQAPG-----APTLERVDVVQPVTFAVMVSLARVWQHHGVTPQAVVGHSQGEIAAAYVAGALSLDDAARVVTLRSKSIAAhLAGKGGMLSLALSEDAVLERLAGFD-GLSVAAVNGPTATVVSGDPVQIEELARACEADGVRARVIPVDYASHSRQVEIIESELAEVLAGLSPQAPRVPFFSTLEGAWITE-PVLDGGYWYRNLRHRVGFAPAVETLATDEGFTHFVEVSAHPVLTMALPGTV-----------TGLATLRRD 925
8*************************************************7.....********************************************************************988***********************96.************************************************************************************.*************************9666***************9988885...........557777775 PP
You may also want to see where the domains are located in the input sequence; using the DNA feature viewer developed by the Edinburgh Genome Foundry, we can build a summary graph aligning the protein sequences to the same reference axis:
[7]:
from dna_features_viewer import GraphicFeature, GraphicRecord
import matplotlib.pyplot as plt
# create an index so we can retrieve a Sequence from its name
seq_index = { seq.name:seq for seq in sequences }
fig, axes = plt.subplots(nrows=len(hits), figsize=(16, 6), sharex=True)
for ax, hit in zip(axes, hits):
# add one feature per domain
features = [
GraphicFeature(start=d.alignment.target_from-1, end=d.alignment.target_to)
for d in hit.domains
]
length = len(seq_index[hit.name])
desc = seq_index[hit.name].description
# render the feature records
record = GraphicRecord(sequence_length=length, features=features)
record.plot(ax=ax)
ax.set_title(desc)
# make sure everything fits in the final graph!
fig.tight_layout()
Matplotlib is building the font cache; this may take a moment.

Checking individual positions for catalytic activity#
First let’s define a function to iterate over an alignement; this will come in handy later. This function yields the position in the alignment (using the HMM coordinates) and the aligned amino acid, skipping over gaps in the HMM sequence.
[8]:
def iter_target_match(alignment):
position = alignment.hmm_from
for hmm_letter, amino_acid in zip(alignment.hmm_sequence, alignment.target_sequence):
if hmm_letter != ".":
yield position, amino_acid
position += 1
Now, for the final step, we want to check for the specificity of the enzyme domains; Del Vecchio et al. have identified two amino acids in the acyltransferase domain that once muted will decide of the enzyme specificity for either malonyl-CoA or methylmalonyl-CoA:
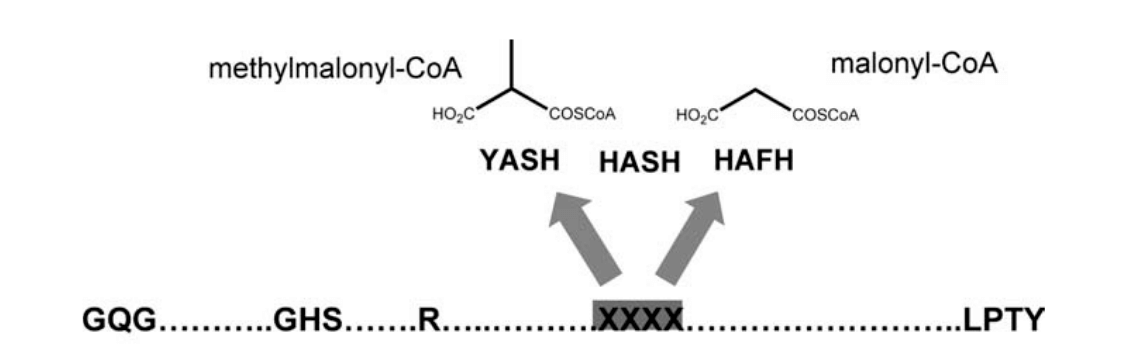
For this, we need to check the alignment produced by HMMER, and verify the residues of the catalytic site correspond to the ones expected by the authors. We use the function we defined previously, first to check the core amino acids are not muted, and then to check the specificity of the two remaining residues.
[9]:
POSITIONS = [ 93, 94, 95, 120, 196, 198]
EXPECTED = ['G', 'H', 'S', 'R', 'A', 'H']
SPECIFICITY = [195, 197]
for hit in hits:
print("\nIn sequence {!r}:".format(hit.name))
for domain in hit.domains:
ali = domain.alignment
aligned = dict(iter_target_match(ali))
print("- Found PKSI-AT domain at positions {:4} to {:4}".format(ali.target_from, ali.target_to))
try:
signature = [ aligned[x] for x in POSITIONS ]
spec = [ aligned[x] for x in SPECIFICITY ]
except KeyError:
print(" -> Domain likely too short")
continue
if signature != EXPECTED:
print(" -> Substrate specificity unknown")
elif spec == ["H", "F"]:
print(" -> Malonyl-CoA specific")
elif spec == ["Y", "S"]:
print(" -> Methylmalonyl-CoA specific")
else:
print(" -> Neither malonyl-CoA nor methylmalonyl-CoA specific")
In sequence 'sp|Q9ZGI5|PIKA1_STRVZ':
- Found PKSI-AT domain at positions 635 to 925
-> Methylmalonyl-CoA specific
- Found PKSI-AT domain at positions 1651 to 1927
-> Methylmalonyl-CoA specific
- Found PKSI-AT domain at positions 3181 to 3475
-> Malonyl-CoA specific
In sequence 'sp|Q9ZGI2|PIKA4_STRVZ':
- Found PKSI-AT domain at positions 563 to 837
-> Methylmalonyl-CoA specific
In sequence 'sp|A0A089QRB9|MSL3_MYCTU':
- Found PKSI-AT domain at positions 540 to 834
-> Neither malonyl-CoA nor methylmalonyl-CoA specific
In sequence 'sp|Q9Y8A5|LOVB_ASPTE':
- Found PKSI-AT domain at positions 562 to 585
-> Domain likely too short
- Found PKSI-AT domain at positions 651 to 854
-> Neither malonyl-CoA nor methylmalonyl-CoA specific
In sequence 'sp|Q54FI3|STLB_DICDI':
- Found PKSI-AT domain at positions 625 to 726
-> Domain likely too short
- Found PKSI-AT domain at positions 766 to 838
-> Domain likely too short
- Found PKSI-AT domain at positions 880 to 944
-> Domain likely too short